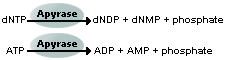In chain terminator sequencing (Sanger sequencing), extension is
initiated at a specific site on the template DNA by using a short
oligonucleotide 'primer' complementary to the template at that region.
The oligonucleotide primer is extended using a DNA polymerase,
an enzyme that replicates DNA. Included with the primer and DNA
polymerase are the four deoxynucleotide bases (DNA building blocks),
along with a low concentration of a chain terminating nucleotide (most
commonly a di-deoxynucleotide). Limited incorporation of the
chain terminating nucleotide by the DNA polymerase results in a series
of related DNA fragments that are terminated only at positions where
that particular nucleotide is used. The fragments are then
size-separated by electrophoresis in a slab polyacrylamide gel, or more
commonly now, in a narrow glass tube (capillary) filled with a viscous
polymer.
An alternative to the labelling of the primer is to label the
terminators instead, commonly called 'dye terminator sequencing'. The
major advantage of this approach is the complete sequencing set can be
performed in a single reaction, rather than the four needed with the
labeled-primer approach. This is accomplished by labelling each of the
dideoxynucleotide chain-terminators with a separate fluorescent dye,
which fluoresces at a different wavelength.
This method is easier and quicker than the dye primer approach, but may
produce more uneven data peaks (different heights), due to a template
dependent difference in the incorporation of the large dye
chain-terminators. This problem has been significantly reduced with the
introduction of new enzymes and dyes that minimize incorporation
variability.
This method is now used for the vast majority of sequencing
reactions as it is both simpler and cheaper. The major reason for this
is that the primers do not have to be separately labelled (which can be
a significant expense for a single-use custom primer), although this is
less of a concern with frequently used 'universal' primers.
>> 처음 생긴 시퀀싱 방법으로, 많이 알려진 방법이고, 현재 학교에서 배우는게 이 방법이다. chain termination method라고 하여, 이름처럼 중간중간 끊어진 부분을 읽는 것이다. PCR할 때 dNTP와 함께 약간의 ddNTP를 첨가하여 사슬에 ddNTP가 결합하는 부분은 더이상 진행되지 않게 된다. 무작위적으로 ddNTP가 결합하면서 다른 길이의 DNA 사슬이 중합되고, 전기영동을 통해서 분리해내면 길이 순서대로 정렬된다. ddNTP에는 형광 dye가 결합되어 있어서, A T G C 가 염기를 구분할 수 있다. 따라서, 이를 전기영동하고 형광dye를 순서대로 읽으면 해당 DNA의 서열을 알 수 있게 된다. 우리 실험실에 있는 ABI사의 sequencing 장비가 이 원리를 이용한다.
During capillary electrophoresis, the products of the
cycle sequencing reaction are injected electrokinetically into
capillaries filled with polymer. High voltage is applied so that the
negatively charged DNA fragments move through the polymer in the
capillaries toward the positive electrode.
A high voltage is applied so that the negatively
charged DNA fragments move through the polymer in the capillaries
toward the positive electrode (Figure 1). Capillary electrophoresis can
resolve DNA molecules that differ in molecular weight by only one
nucleotide.
Figure 1: Fluorescently labeled DNA fragments move through a capillary
Figure 2: DNA fragments pass through a laser beam and optical detector
Shortly before reaching the positive electrode,
the fluorescently labeled DNA fragments, separated by size, move
through the path of a laser beam. The laser beam causes the dyes on the
fragments to fluoresce. An optical detection device on Applied
Biosystems DNA analyzers detects the fluorescence (Figure 2). The Data
Collection Software converts the fluorescence signal to digital data,
then records the data in a *.ab1 file. Because each dye emits light at
a different wavelength when excited by the laser, all four colors, and
therefore, all four bases, can be detected and distinguished in one
capillary injection.
After electrophoresis, data collection software
creates a sample file of the raw data. Using downstream software
applications, further data analysis is required to translate the
collected color-data images into the corresponding nucleotide bases.
Primary Analysis
These tools convert the images gathered during Data Collection into
all four colors, representing the four corresponding nucleotide bases
(Figure 1). For example, our Sequence Analysis Software is a primary
analysis tool that must be used after collection is completed. The
Sequence Analysis software application allows users to basecall and
re-basecall, trim data ends, display, edit and print sample files.
Primary analysis software processes the your raw data in an *.ab1 file
using algorithms and applies the following analysis settings to the
results:
Basecalling The selected basecaller processes the fluorescence signals,
then assigns a base to each peak (A, C, G, T, or N). If the KB™
basecaller is used, it also provides per-base quality value
predictions, optional mixed base calling, and automatic identification
of failed samples.
Figure 1: Primary Analysis Software results display each of the 4 bases as a different color
Mobility Shift Correction The mobility file compensates for the change in DNA fragment
mobility caused by the dye molecule attached to the DNA fragment and
changes the color designation of bases depending on the type of
chemistry used to label the DNA.
Quality Value (QV) If the KB basecaller is used for analysis, the software
assigns a QV for each base. The QV predicts the probability of a
basecall error. For example, a QV of 20 predicts an error rate of 1%.
The quality prediction algorithm is calibrated to return QVs that
conform to the industry-standard relationship established by the Phred
software. If your pipeline involves analysis with Phred software to
assign QVs after the data is basecalled, you can simplify your workflow
and use the KB basecaller instead. The KB basecaller can perform
basecalling and assign QVs. Then, you can generate *phd.1 or *.scf
files using the KB basecaller to integrate with your downstream
pipeline.
Secondary Analysis
These tools allow you to further refine your results. Algorithms in
our secondary analysis software products perform a number of functions
supporting applications such as mutation detection and genotyping, and
produce graphical outputs.
Pyrosequencing,
which was originally developed by Mostafa Ronaghi, has been
commercialized by Biotage (for low throughput sequencing) and 454 Life
Sciences (for high-throughput sequencing). The latter platform
sequences roughly 100 megabases in a 7-hour run with a single machine.
In the array-based method (commercialized by 454 Life Sciences),
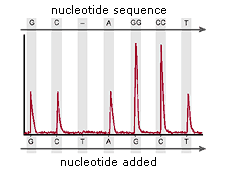
single-stranded DNA is annealed to beads and amplified via emPCR. These DNA-bound beads are then placed into wells on a fiber-optic chip along with enzymes which produce light in the presence of ATP.
When free nucleotides are washed over this chip, light is produced as
ATP is generated when nucleotides join with their complementary base pairs.
Addition of one (or more) nucleotide(s) results in a reaction that
generates a light signal that is recorded by the CCD camera in the
instrument. The signal strength is proportional to the number of
nucleotides, for example, homopolymer stretches, incorporated in a
single nucleotide flow.
>> Pyrosequencing은 최근에 새롭게 등장한 방법이다. 좀 복잡하다.;;; 나도 Sanger 말고 이런 방법도 있다는걸 안게 얼마 안되었다. Sanger sequencing이 한번에 sequencing할 수 있는 길이가 짧아서 게놈 단위의 분석이 어려운데 비해서 이 방법은 하나의 장비에서 단 7시간동안 1억bp나 되는 긴 서열의 분석이 가능하다.
>> Pyrosequencing은 4가지 효소인 DNA polymerase, Sulfurylase, Luciferase, Apyrase등의 Enzyme Cascade를 응용한 것으로 그 원리는 다음과 같다. 우선 Sequencing Primer 가 분석 하려는 DNA 가닥에 결합한다. 그 후 특정의 염기가 반응용액에 떨어지면 DNA 염기 중합반응이 일어나면서 Pyrophosphate(PPi)기가 떨어져 나온다. 이때 Pyrophosphate는 Sulfurylase에 의해 APS(adensosine 5'' phosphosulfate)와 반응하여 ATP를 만들어내고, 이 ATP는 Luciferase를 활성화 하여 Luciferin을 Oxyluciferin으로 산화 시킨다. 이때 Oxyluciferin이 빛을 내게 되며, 이 빛을 CCD camera로 검출하게 되며, 이에 따라 특정 염기를 인식하여 분석을 한다. (출처:(주)BMS 자료)
<Procedeure>
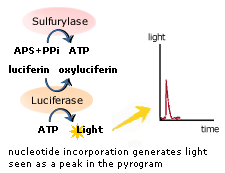
The method is based on detecting the activity of DNA polymerase with a chemiluminescentenzyme. Essentially, the method allows sequencing of a single strand of DNA
by synthesizing the complementary strand along it, one base pair at a
time, and detecting which base was actually added at each step. The
template DNA is immobilized, and solutions of A, C, G, and T nucleotides are added sequentially. Light is produced only when the nucleotide
solution complements the first unpaired base of the template. The
sequence of solutions which produce chemiluminescent signals allows the
determination of the sequence of the template. ssDNA template is hybridized to a sequencing primer and incubated with the enzymes DNA polymerase, ATP sulfurylase, luciferase and apyrase, and with the substrates adenosine 5´ phosphosulfate (APS) and luciferin.
The addition of one of the four deoxynucleotide triphosphates (dNTPs)(in
the case of ATP we add ATPαS which is not a substrate for a luciferase)
initiates the second step. DNA polymerase incorporates the correct,
complementary dNTPs onto the template. This incorporation releases pyrophosphate (PPi) stoichiometrically.
ATP sulfurylase quantitatively converts PPi to ATP
in the presence of adenosine 5´ phosphosulfate. This ATP acts as fuel
to the luciferase-mediated conversion of luciferin to oxyluciferin that generates visible light in amounts that are proportional to the amount
of ATP. The light produced in the luciferase-catalyzed reaction is
detected by a camera and analyzed in a program.
Unincorporated nucleotides and ATP are degraded by the apyrase, and the reaction can restart with another nucleotide.
Currently, a limitation of the method is that the lengths of
individual reads of DNA sequence are in the neighborhood of 300-500
nucleotides, shorter than the 800-1000 obtainable with chain termination methods (e.g. Sanger sequencing). This can make the process of genome assembly more difficult, particularly for sequence containing a large amount of repetitive DNA.
As of 2007, pyrosequencing is most commonly used for resequencing or
sequencing of genomes for which the sequence of a close relative is
already available.
The templates for pyrosequencing can be made both by solid phase
template preparation (Streptavidin coated magnetic beads) and enzymatic
template preparation (Apyrase+Exonuclease).