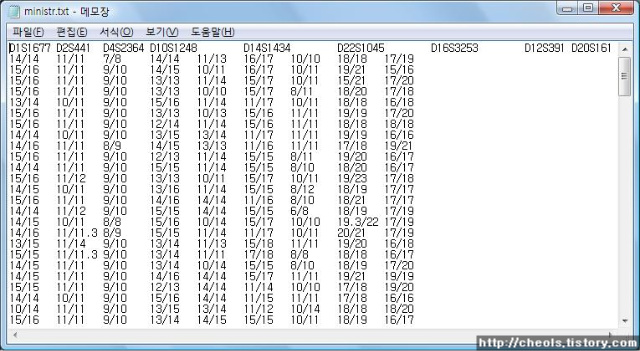
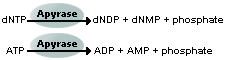동북공정의 연구물인 ‘고대 중국 고구려 역사 속론’(2003년)에는 고구려인이 중국의 고대 국가인 은나라와 상나라의 씨족에서
분리됐다고 주장하고 있다. 한국인과 중국 한족은 혈연적으로 한 핏줄이란 얘기인데, 과연 그럴까?
2003년 단국대 생물과학과 김욱 교수는 동아시아인 집단에서 추출한 표본을 대상으로 부계를 통해 유전되는 Y염색체의
유전적 변이를 분석했다. 이 결과 한국인은 주로 몽골과 동․남부 시베리아인에게서 흔히 볼 수 있는 유전자 형, 그리고 동남아시아
및 중국 남․북부에서 흔히 볼 수 있는 유전자형이 모두 발견되었다.
한국인은 동아시아의 여러 민족 가운데서 동남아시아인인 중국 동북부 만주족과 유전적으로 가장 유사했고, 중국 묘족이나
베트남 등 일부 동남아시아인과도 비슷했다. 이는 한민족이 크게 북방계와 남방계의 혼합 민족이라는 사실을 보여준다. 2300여 년
전 농경문화와 일본어를 전달한 야요이족이 한반도를 거쳐 일본 본토로 이주했음을 나타내는 유전학적 증거이기도 하다.
2006년 김 교수는 모계유전을 하는 미토콘드리아 DNA도 분석했다. Y염색체가 아버지를 통해 아들에게만 전달되는 부계유전을 하는 것과 달리 미토콘드리아 DNA는 어머니를 통해 아들과 딸 모두에게 전달된다. 더욱이 미토콘드리아 DNA는 돌연변이율이 높고, 교차가 일어나지 않는다는 장점이 있다. 이 때문에 인류의 진화과정에서 일어나는 돌연변이 정보인 하플로타입 상태를 분석해 조상을 추적해 낼 수 있다.
하플로타입이란 일련의 특이한 염기서열이나 여러 유전자들이 가깝게 연관돼 한 단위로 표시될 수 있는 유전자형을 가리킨다. 하플로그룹은 같은 미토콘드리아 DNA 유전자형을 가진 그룹으로 보면 된다. 한국인은 3명 가운데 1명꼴로 몽골과 중국 중북부의 동북아시아에 많이 분포하는 하플로그룹D 계통이 가장 많았고, 전체적으로 한국인의 60% 가량이 북방계로, 40% 가량이 남방계로 분류됐다.
유전적인 분화 정도를 통해 분석한 결과, 한국인은 중국 조선족과 만주족 그리고 일본인 순으로 가까웠다. 그러나 중국
한족은 베트남과 함께 다른 계통에 묶여 한국인과는 유전적으로 다소 차이를 보였다. 동북아시아에 속한 중국 북경의 한족은 한국인과
다소 비슷한 결과를 보였지만 중국 남방의 한족과는 유전적으로 차이가 있었다.
특히 만주족과 중국 동북 3성인 랴오닝(遼寧)·지린(吉林)·헤이룽장(黑龍江)에 살고 있는 조선족은 중국 한족보다는
한국인과 유전적으로 더 가까웠다. 이 때문에 김 교수는 “과거 한반도와 만주 일대에서 활동했던 고구려인의 유전적 특성은 중국
한족 집단보다 한국인 집단에 더 가깝다”고 밝혔다.
이와 함께 최근 역사학계에서는 중국 한족을 물리치고 중원을 점령했던 금나라의 여진족(훗날 만주족)이 신라인의 후예라는
주장이 제기되고 있다. 금나라의 역사를 기록한 금사(金史)에는 “금태조가 고려에서 건너온 함보를 비롯한 3형제의 후손이다”는
대목이 나온다. 또 금을 계승한 청나라의 건륭제 때 집필된 ‘흠정만루원류고’에는 금나라의 명칭이 신라 김(金)씨에서 비롯됐다는
내용도 등장한다.
<한국인의 유전체 염기서열을 분석해 보면 우리의 유전자가 누구와 가까운지 알
수 있다. 사진은 생명공학기업인 마크로젠이 소개한 한국인 유전자 지도 초안
이다. 사진 제공. 동아일보>
청나라 황실의 만주어성 ‘아이신줴뤄’ 중 씨족을 가리키는 아이신은 금(金)을 뜻한다. 이는 아이신줴뤄를 한자로 가차한 애신각라(愛新覺羅)에 “신라(新羅)를 사랑하고, 기억하자”는 뜻이 담겼다는 가설을 뒷받침한다.
이런 결과로 볼 때 한국인의 유전자는 북방계가 다소 우세하지만 남방계와 북방계의 유전자가 복합적으로 섞여있다.
4000~5000년 동안 한반도와 만주 일대에서 동일한 언어와 문화를 발달시키고 역사적인 경험을 공유하면서 유전적으로 동질성을
갖는 한민족으로 발전했던 것으로 보인다.
따라서 만주에 살던 이들은 중국 황하 유역을 중심으로 발원한 한족과는 달리 한반도에 살던 이들과 깊은 혈연관계였음을
추정해 볼 수 있다. 나아가 금나라와 청나라를 세웠던 여진족과 만주족의 역사를 한국사에 새로 편입시켜야 할지도 모를 일이다.
우리는 흔히 스스로 ‘단일민족’이라고 말한다. 여기서 단일민족은 오랜 세월을 거치면서 유전적 동질성을 획득했다는 의미이지
한국인의 기원이 하나라는 의미는 아니다. 오히려 한국인은 동아시아 내에서 남방과 북방의 유전자가 복합적으로 이뤄져 형성된,
다양성을 지닌 민족이다.
유전적으로 다양한 집단은 그렇지 않은 집단에 비해 집단 구성원이 갖고 있는 유전적 다양성이 세대를 통해 유지될 확률이 크다. 그리고 집단의 안정성도 높아진다.
다양한 유전자를 보유한 집단은 단순한 집단에 비해 집단이 유지되고 진화하는데 유리하다는 뜻이다. 이런 의미에서 한국인은
‘잡종강세’의 전형적인 집단이다. 어쩌면 중국이 동북공정을 서두르는 이유도 한국인의 유전적 다양성을 두려워해서가 아닐까?
글 : 서금영 과학칼럼니스트
KISTI NDSL(과학기술정보통합서비스) 지식링크
○관련 논문 정보
Y-염색체 DNA haplogroup과 동아시아인집단에서 초기농경민족의 집단팽창[바로가기]
인간 Y 염색체: 구조, 기능 그리고 진화[바로가기]
한국인 집단의 미토콘드리아 DNA HV1 부위에서의 염기서열 다양성[바로가기]
○관련 특허 정보
미토콘드리아 유전자 정보를 이용한 개인인식표지(한국등록특허)[바로가기]
인간 미토콘드리아 DNA의 변이 판별 방법과 변이 판별용폴리뉴클레오티드 프로브, DNA 칩 및 키트(한국등록특허)[바로가기]
유전자의 변이 분석 키트(한국등록특허)[바로가기]
○해외 동향분석 자료
차세대 염기서열 분석기술과 돌연변이 - 2009년 [바로가기]
네안데르탈인 유전체 분석완료 - 2009년 [바로가기]
사람의 미토콘드리아 DNA 점 돌연변이 병리학의 20년 - 2009년 [바로가기]
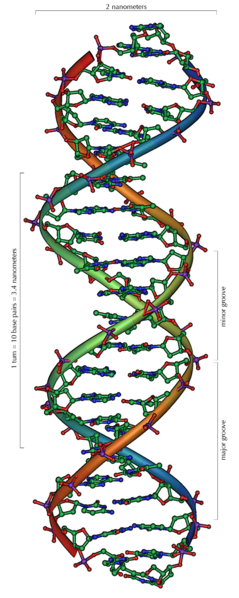
DNA (Deoxyribonucleic acid) : 꼬인 사다리처럼 보이는 긴 분자. 네 종류의 간단한 단위로 구성되고, 문자를 이용해 책에 정보를 지니는 것처럼, 이들 단위의 서열이 정보를 지닌다. [A long molecule
that looks like a twisted ladder. It is made of four types of simple
units and the sequence of these units carries information, just as the
sequence of letters carries information on a page.]
Nucleotides : DNA 사다리의 가로대를 형성하며, DNA안에서 반복되는 단위이다. 네 종류의 뉴클레오티드(A, T, G, C)가 있고 이들 뉴클레오티드의 서열이 정보를 지닌다. [They form the rungs
of the DNA ladder and are the repeating units in DNA. There are four
types of nucleotides (A, T, G and C) and it is the sequence of these
nucleotides that carries information.]
Chromosome : 염색체. 세포에서 DNA를 운반하기 위해 포장된 것. 하나의 긴 DNA조각을 포함하여 치밀한 구조로 서로 감기고 다발로 묶여있다. 종이 다른 식물과 동물들은 염색체의 갯수와 크기가 서로 다르다. [A package for carrying DNA in the cells. They contain a single long
piece of DNA that is wound up and bunched together into a compact
structure. Different species of plants and animals have different
numbers and sizes of chromosomes.]
Gene : 유전자. DNA의 조각 또는 단편. 유전자는 뉴클레오티드라는 알파벳의 문자로 이루어진 문장과 같으며, 그들 유전자 사이에서 생명체의 물리적 발달과 행동을 지시한다. 유전자는 조리법 또는 사용설명서와 같아서, 생명체가 필요로하는 어떤것 - 눈 또는 다리 또는 상처 치료와 같은 것을 만들거나 하는데 필요한 정보를 제공한다. [A segment of DNA.
Genes are like sentences made of the "letters" of the nucleotide
alphabet, between them genes direct the physical development and
behavior of an organism. Genes are like a recipe or instruction book,
providing information that an organism needs so it can build or do
something - like making an eye or a leg, or repairing a wound.]
Allele : 대립인자. 생명체가 지니고 있는 주어진 유전자의 다른 형태. 예를 들어, 인간에서 눈 색 유전자의 한 대립인자는 녹색 눈을 만들고, 다른 눈 색 유전자의 대립인자는 갈색 눈을 만든다. [The different forms of a given gene that an organism may possess.
For example, in humans, one allele of the eye-color gene produces green
eyes and another allele of the eye-color gene produces brown eyes.]
Genome : 특정 생명체에 있는 유전자의 완전한 세트. [The complete set of genes in a particular organism.]
Genetic engineering : 사람들이 새로운 유전자를 추가하거나 게놈으로부터 유전자를 제거하여 생명체를 변화시킬 때 유전공학이라 한다. [When people change an organism by adding new genes, or deleting genes from its genome.]
Mutation : 유전자의 DNA 서열에 변화가 일어난 사건. [An event that changes the sequence of the DNA in a gene.]
생물학의 분야인 유전학(genetics)은 살아있는 생명체의 유전(heredity)과 변이(variation)의 과학이다. 역사이전 시대부터 살아있는 것은 그들의 부모로부터 특성을 물려받는다는 사실을 알고 선택적 교배를 통해서 작물과 동물을 향상시켰다. 그러나, 유전학의 새로운 과학은 유전의 과정을 이해하는 것으로, 19세기 중반에 그레고르 멘델(Gregor Mendel)에 의해 처음 시작되었다. 비록 멘델은 유전에 대한 물리적인 기본을 알지 못했지만, 그는 생명체의 유전 특성이 분리되어 있다는 특성을 발견했다. 이들 기본적인 유전의 단위를 오늘날 유전자(gene)라 부른다.
유전학은 유전의 기본 단위인 유전자의 연구를 통해서 유전, 유전적 변이 그리고 선택적 교배를 다루는 생물학의 한 가지이다. 유전자는 네 종류의 뉴클레오티드(nucleotide)의 사슬로 구성된 분자인 DNA안에 상응하는 영역이다. - 이들 뉴클레오티드의 서열은 생명체가 물려받는 유전적 정보이다. DNA는 기본적으로 서로 상보성(complementary)을 띠는 각 사슬의 뉴클레오티드와 함께 이중 가닥 형태를 나타낸다. 각 사슬은 새로운 파트너 가닥을 만드는 주형(template)으로 작용할 수 있다. - 이것은 유전자의 사본을 만들어서 유전될 수 있도록 하는 물리적 방법이다.
유전자에 있는 뉴클레오티드의 서열은 세포에 의해 번역되어 아미노산의 사슬을 생산하고 단백질을 만든다. - 단백질에 있는 아미노산의 순서는 유전자에 있는 뉴클레오티드의 순서와 대응한다. 이것은 유전 암호(genetic code)라고 알려져 있다. 단백질 내의 아미노산은 3차원 형태로 접힘(fold)으로써 정의된다.; 이 구조는 단백질이 기능을 나타낼 수 있도록 한다. 단백질은 세포가 살아가기 위해 필요한 거의 모든 기능을 수행한다. 유전자에 있는 DNA의 변화는 단백질의 아미노산에 변화를 일으킬 수 있고, 단백질의 형태와 기능을 변화시킨다.; 이것은 세포와 전체 생명체에 극적인 영향을 줄 수 있다.
비록 유전학이 생명체의 행동을 나타내는데 큰 역할을 하지만, 그것은 생명체가 경험한 것과 함께 유전학이 결합하여서 궁극적인 결과를 정한다. 예를 들어, 유전자가 사람의 키, 영양과 건강을 정하는 역할을 하지만, 어린시절의 경험 또한 큰 영향을 주게 된다.
유전학(genetics)은 살아있는 생명체가 어떻게 그들 선조의 수많은 특성을 전달하는지에 대해 연구한다. - 예를 들면, 아이들이 보통 그들 가족의 다른 사람들처럼 보고 행동하는 것과 같은 것이다. 유전학은 유전된 특성을 확인하기 위해 노력하고, 구체적으로 어떻게 이들 특성이 세대에서 세대로 전달되는지 연구한다.
유전학에서, 생명체의 특성을 "형질(trait)"이라 부른다. 몇몇 형질은 생명체의 물리적 외형의 특성들이다. 사람의 눈 색, 키 또는 몸무게 등을 예로 들 수 있다. 많은 다른 형태의 형질과 질병에 저항하는 반응 양상의 범위가 존재한다. 형질은 종종 유전된다. 예를 들면, 키크고 날씬한 사람은 키크고 날씬한 자녀를 갖는 경향이 있다. 다른 형질은 유전된 특성과 환경 사이의 상호작용으로부터 나타난다. 예를 들어, 아이가 키가 큰 경향으로 유전되었지만 만약 그가 사는 곳에 음식이 적고 궁핍하게 자랐다면, 키가 작을 것이다. 유전학과 환경이 상호작용하여 형질을 만드는 과정은 이해하기 어려울 수 있다. : 예를 들면, 누군가가 암 또는 심장병으로 죽을 확률은 그들의 가족력과 생활양식 두가지에 의존해서 판단한다.
유전 정보는 DNA라 부르는 긴 분자에 의해 운반되고 이 DNA는 세대를 거쳐서 복제되고 전달된다. 형질은 생명체를 구성하고 운영하기 위한 지시사항으로써 DNA로 운반된다. 이들 지시사항은 유전자라 부르는 DNA의 조각에 포함되어 있다. DNA는 유전 암호에 있는 지시사항을 한자한자 읽어가는 이들 단위의 상태와 함께 간단한 단위의 서열로 구성된다. 이것은 단어를 한자한자 나열한 편지의 상태와 유사하다. 생명체는 이러한 단위의 서열을 읽고 지시사항을 해독한다.
특정 지시사항에 대해 존재하는 유전자가 똑같지는 않다. 유전자의 한 유형의 다른 형태를 그 유전자의 다른 대립인자(allele)라고 부른다. 그 예로써, 머리 색에 대한 한 유전자의 한 대립인자는 검은 머리카락에서 많은 색소를 생산하는 지시사항을 운반하는 반면, 다른 대립인자는 이 지시사항의 잘못된 버전을 줄 수 있고, 그래서 색소를 생산하지 않고 머리카락이 하얗게 된다. 돌연변이(Mutation)는 유전자의 서열이 변하고 따라서 새로운 대립인자가 생기는 무작위적인 사건이다. 돌연변이는 검은 머리카락에 대한 대립인자를 흰 머리카락에 대한 대립인자로 바꾸는 것과 같이 새로운 형질을 만들 수 있다. 새로운 형질의 출현은 진화에서 중요하다.
DNA는 'Deoxyribonucleic acid'의 줄임말로, 현재 알려진 모든 생명체와 일부 바이러스에서 발생과 기능을 위해 이용되는 유전적 명령을 담고있는 '핵산' 입니다. DNA 분자의 주요 역할은 정보를 장기간 저장하는 것입니다. DNA는 단백질과 RNA분자와 같은 세포의 구성요소를 형성하는데 필요한 명령을 갖고있기 때문에, 종종 청사진이나 조리법과 비교됩니다. DNA 조각은 유전자라 부르는 유전 정보를 운반합니다. 그러나 다른 DNA 서열은 구조적인 목적이나 유전 정보의 사용을 조절하기 위해서 존재합니다.
화학적으로, DNA는 당(sugar)으로 구성된 뼈대와 에스테르 결합으로 결합하고 있는 인산기(phosphate group), 그리고 뉴클레오티드(nucleotide)라 부르는 간단한 단위의 긴 중합체(polymer) 두개로 구성됩니다. 이들 두 사슬은 서로 반대방향으로 진행하기 때문에 역평행(anti-parallel)이라고 합니다. 각 당에는 염기(base)라 부르는 4종류의 분자중 하나가 붙어 있습니다. 뼈대를 따라 있는 이들 네 종류 염기의 순서가 정보를 암호화합니다. 이 정보는 단백질 내의 아미노산 순서를 정하는 유전적 코드를 사용해 읽게 됩니다. 코드는 DNA에서 관련된 핵산 RNA로 복사됨으로써 읽혀지고, 이 과정을 전사(transcription)라고 합니다.
세포 내에서, DNA는 염색체(chromosomes)라 부르는 구조로 조직화되어 있습니다. 이들 염색체는 세포가 분열하기 전에 DNA 복제(replication)이라는 과정에 의해서 두배로 복제됩니다. 진핵생물(Eucaryotic organisms, 동물.식물.균류)은 세포 핵 안에 DNA를 저장하지만, 원핵생물(Prokaryotes, 박테리아. 시원생물)은 세포의 세포질(cytoplasm)에서 발견됩니다. 염색체 내에서, 히스톤(histone)과 같은 염색질(chromatin)단백질 이 DNA를 밀집하고 조직화합니다. 이러한 밀집 구조는 DNA와 다른 단백질 사이의 상호작용을 안내하고, DNA 일부분의 전사 조절을 도와줍니다.
In chain terminator sequencing (Sanger sequencing), extension is
initiated at a specific site on the template DNA by using a short
oligonucleotide 'primer' complementary to the template at that region.
The oligonucleotide primer is extended using a DNA polymerase,
an enzyme that replicates DNA. Included with the primer and DNA
polymerase are the four deoxynucleotide bases (DNA building blocks),
along with a low concentration of a chain terminating nucleotide (most
commonly a di-deoxynucleotide). Limited incorporation of the
chain terminating nucleotide by the DNA polymerase results in a series
of related DNA fragments that are terminated only at positions where
that particular nucleotide is used. The fragments are then
size-separated by electrophoresis in a slab polyacrylamide gel, or more
commonly now, in a narrow glass tube (capillary) filled with a viscous
polymer.
An alternative to the labelling of the primer is to label the
terminators instead, commonly called 'dye terminator sequencing'. The
major advantage of this approach is the complete sequencing set can be
performed in a single reaction, rather than the four needed with the
labeled-primer approach. This is accomplished by labelling each of the
dideoxynucleotide chain-terminators with a separate fluorescent dye,
which fluoresces at a different wavelength.
This method is easier and quicker than the dye primer approach, but may
produce more uneven data peaks (different heights), due to a template
dependent difference in the incorporation of the large dye
chain-terminators. This problem has been significantly reduced with the
introduction of new enzymes and dyes that minimize incorporation
variability.
This method is now used for the vast majority of sequencing
reactions as it is both simpler and cheaper. The major reason for this
is that the primers do not have to be separately labelled (which can be
a significant expense for a single-use custom primer), although this is
less of a concern with frequently used 'universal' primers.
>> 처음 생긴 시퀀싱 방법으로, 많이 알려진 방법이고, 현재 학교에서 배우는게 이 방법이다. chain termination method라고 하여, 이름처럼 중간중간 끊어진 부분을 읽는 것이다. PCR할 때 dNTP와 함께 약간의 ddNTP를 첨가하여 사슬에 ddNTP가 결합하는 부분은 더이상 진행되지 않게 된다. 무작위적으로 ddNTP가 결합하면서 다른 길이의 DNA 사슬이 중합되고, 전기영동을 통해서 분리해내면 길이 순서대로 정렬된다. ddNTP에는 형광 dye가 결합되어 있어서, A T G C 가 염기를 구분할 수 있다. 따라서, 이를 전기영동하고 형광dye를 순서대로 읽으면 해당 DNA의 서열을 알 수 있게 된다. 우리 실험실에 있는 ABI사의 sequencing 장비가 이 원리를 이용한다.
During capillary electrophoresis, the products of the
cycle sequencing reaction are injected electrokinetically into
capillaries filled with polymer. High voltage is applied so that the
negatively charged DNA fragments move through the polymer in the
capillaries toward the positive electrode.
A high voltage is applied so that the negatively
charged DNA fragments move through the polymer in the capillaries
toward the positive electrode (Figure 1). Capillary electrophoresis can
resolve DNA molecules that differ in molecular weight by only one
nucleotide.
Figure 1: Fluorescently labeled DNA fragments move through a capillary
Figure 2: DNA fragments pass through a laser beam and optical detector
Shortly before reaching the positive electrode,
the fluorescently labeled DNA fragments, separated by size, move
through the path of a laser beam. The laser beam causes the dyes on the
fragments to fluoresce. An optical detection device on Applied
Biosystems DNA analyzers detects the fluorescence (Figure 2). The Data
Collection Software converts the fluorescence signal to digital data,
then records the data in a *.ab1 file. Because each dye emits light at
a different wavelength when excited by the laser, all four colors, and
therefore, all four bases, can be detected and distinguished in one
capillary injection.
After electrophoresis, data collection software
creates a sample file of the raw data. Using downstream software
applications, further data analysis is required to translate the
collected color-data images into the corresponding nucleotide bases.
Primary Analysis
These tools convert the images gathered during Data Collection into
all four colors, representing the four corresponding nucleotide bases
(Figure 1). For example, our Sequence Analysis Software is a primary
analysis tool that must be used after collection is completed. The
Sequence Analysis software application allows users to basecall and
re-basecall, trim data ends, display, edit and print sample files.
Primary analysis software processes the your raw data in an *.ab1 file
using algorithms and applies the following analysis settings to the
results:
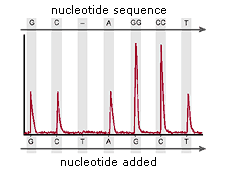
Basecalling The selected basecaller processes the fluorescence signals,
then assigns a base to each peak (A, C, G, T, or N). If the KB™
basecaller is used, it also provides per-base quality value
predictions, optional mixed base calling, and automatic identification
of failed samples.
Figure 1: Primary Analysis Software results display each of the 4 bases as a different color
Mobility Shift Correction The mobility file compensates for the change in DNA fragment
mobility caused by the dye molecule attached to the DNA fragment and
changes the color designation of bases depending on the type of
chemistry used to label the DNA.
Quality Value (QV) If the KB basecaller is used for analysis, the software
assigns a QV for each base. The QV predicts the probability of a
basecall error. For example, a QV of 20 predicts an error rate of 1%.
The quality prediction algorithm is calibrated to return QVs that
conform to the industry-standard relationship established by the Phred
software. If your pipeline involves analysis with Phred software to
assign QVs after the data is basecalled, you can simplify your workflow
and use the KB basecaller instead. The KB basecaller can perform
basecalling and assign QVs. Then, you can generate *phd.1 or *.scf
files using the KB basecaller to integrate with your downstream
pipeline.
Secondary Analysis
These tools allow you to further refine your results. Algorithms in
our secondary analysis software products perform a number of functions
supporting applications such as mutation detection and genotyping, and
produce graphical outputs.
Pyrosequencing,
which was originally developed by Mostafa Ronaghi, has been
commercialized by Biotage (for low throughput sequencing) and 454 Life
Sciences (for high-throughput sequencing). The latter platform
sequences roughly 100 megabases in a 7-hour run with a single machine.
In the array-based method (commercialized by 454 Life Sciences),
single-stranded DNA is annealed to beads and amplified via emPCR. These DNA-bound beads are then placed into wells on a fiber-optic chip along with enzymes which produce light in the presence of ATP.
When free nucleotides are washed over this chip, light is produced as
ATP is generated when nucleotides join with their complementary base pairs.
Addition of one (or more) nucleotide(s) results in a reaction that
generates a light signal that is recorded by the CCD camera in the
instrument. The signal strength is proportional to the number of
nucleotides, for example, homopolymer stretches, incorporated in a
single nucleotide flow.
>> Pyrosequencing은 최근에 새롭게 등장한 방법이다. 좀 복잡하다.;;; 나도 Sanger 말고 이런 방법도 있다는걸 안게 얼마 안되었다. Sanger sequencing이 한번에 sequencing할 수 있는 길이가 짧아서 게놈 단위의 분석이 어려운데 비해서 이 방법은 하나의 장비에서 단 7시간동안 1억bp나 되는 긴 서열의 분석이 가능하다.
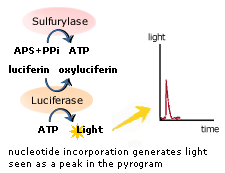
>> Pyrosequencing은 4가지 효소인 DNA polymerase, Sulfurylase, Luciferase, Apyrase등의 Enzyme Cascade를 응용한 것으로 그 원리는 다음과 같다. 우선 Sequencing Primer 가 분석 하려는 DNA 가닥에 결합한다. 그 후 특정의 염기가 반응용액에 떨어지면 DNA 염기 중합반응이 일어나면서 Pyrophosphate(PPi)기가 떨어져 나온다. 이때 Pyrophosphate는 Sulfurylase에 의해 APS(adensosine 5'' phosphosulfate)와 반응하여 ATP를 만들어내고, 이 ATP는 Luciferase를 활성화 하여 Luciferin을 Oxyluciferin으로 산화 시킨다. 이때 Oxyluciferin이 빛을 내게 되며, 이 빛을 CCD camera로 검출하게 되며, 이에 따라 특정 염기를 인식하여 분석을 한다. (출처:(주)BMS 자료)
<Procedeure>
The method is based on detecting the activity of DNA polymerase with a chemiluminescentenzyme. Essentially, the method allows sequencing of a single strand of DNA
by synthesizing the complementary strand along it, one base pair at a
time, and detecting which base was actually added at each step. The
template DNA is immobilized, and solutions of A, C, G, and T nucleotides are added sequentially. Light is produced only when the nucleotide
solution complements the first unpaired base of the template. The
sequence of solutions which produce chemiluminescent signals allows the
determination of the sequence of the template. ssDNA template is hybridized to a sequencing primer and incubated with the enzymes DNA polymerase, ATP sulfurylase, luciferase and apyrase, and with the substrates adenosine 5´ phosphosulfate (APS) and luciferin.
The addition of one of the four deoxynucleotide triphosphates (dNTPs)(in
the case of ATP we add ATPαS which is not a substrate for a luciferase)
initiates the second step. DNA polymerase incorporates the correct,
complementary dNTPs onto the template. This incorporation releases pyrophosphate (PPi) stoichiometrically.
ATP sulfurylase quantitatively converts PPi to ATP
in the presence of adenosine 5´ phosphosulfate. This ATP acts as fuel
to the luciferase-mediated conversion of luciferin to oxyluciferin that generates visible light in amounts that are proportional to the amount
of ATP. The light produced in the luciferase-catalyzed reaction is
detected by a camera and analyzed in a program.
Unincorporated nucleotides and ATP are degraded by the apyrase, and the reaction can restart with another nucleotide.
Currently, a limitation of the method is that the lengths of
individual reads of DNA sequence are in the neighborhood of 300-500
nucleotides, shorter than the 800-1000 obtainable with chain termination methods (e.g. Sanger sequencing). This can make the process of genome assembly more difficult, particularly for sequence containing a large amount of repetitive DNA.
As of 2007, pyrosequencing is most commonly used for resequencing or
sequencing of genomes for which the sequence of a close relative is
already available.
The templates for pyrosequencing can be made both by solid phase
template preparation (Streptavidin coated magnetic beads) and enzymatic
template preparation (Apyrase+Exonuclease).
Y-SNP-genotyping – a new approach in forensic analysis
R. Lessiga, M. Zoledziewskac, K. Fahrb, J. Edelmanna, M. Kostrzewab, T. Doboszc, W.J. Kleemann
Forensic Science International 154 (2005) 128–136
Y-chromosomal DNA polymorphisms, especially Y-STRs are well established in forensic routine case work. The STRs are used for identification in paternity deficiency cases and stain analysis with complicate mixtures of male and female DNA. In contrast, Y-chromosomal SNPs are a new tool in forensic investigations. At present, Y-SNPs are mainly used in molecular anthropology for evolutionary studies. Nevertheless, these markers could also provide very useful information for the analysis of forensic cases. The aim of the presented study was to test Y-SNP-typing for stain analyses using different methods—SNaPshot and MALDI-TOF MS. Both methods are based on the principle of minisequencing. The selected Y-SNP markers are suited to define the most important European haplogroups.
>> Y-STR은 법과학 사건 조사에서 부계 식별 또는 남성과 여성의 DNA가 섞여있을 경우에 유용하게 이용할 수 있다. STR과는 달리, Y-SNP는 법과학 조사에서 새롭게 이용되는 도구이다. 현재는 주로 인류학의 진화 연구에 이용되고 있다. 그렇지만 이들 마커는 이들은 법과학 사건 분석에도 매우 유용한 정보를 제공해 줄 수 있다. 이 연구의 목표는 다른 방법을 사용하여 흔적에 대한 Y-SNP typing 분석방법 두가지(SNaPshot 과 MALDI-TOF MS)를 테스트해보는 것이다. 두 가지 방법은 minisequencing을 기반으로 하고 있다.
==========
* genotyping of Y-SNPs using the genoSNIP assay based on MALDI-TOF MS is less time consuming.
* MALDI-TOF MS is not as widely distributed, the mass spectrometry has the disadvantage of high starting costs so far.
* Y-SNPs correctly also in samples containing less than 125pg DNA.
* High amount of female DNA did not disturb the PCR reaction of the male part.
* detection of the haplogroups can give additional information in the routine of criminalistics to identify an unknown body or perpetrator.
* A special advantage of the SNPs is the very short length of the fragments, what is very important in particular for the analysis of degraded DNA.
<가능한 분석> The analyses Arlequin can
perform on the data fall into two main categories: intra-population and
inter-population methods. In the first category statistical information is
extracted independently from each population, whereas in the second category,
samples are compared to each other.
Intra-population methods:
Short description:
Standard indices
Some diversity measures like the number of
polymorphic sites, gene diversity.
Molecular diversity
Calculates several diversity
indices like nucleotide diversity, different estimators of the population
parameter q.
Mismatch distribution
The distribution of the number of pairwise
differences between haplotypes, from which parameters of a demographic
(NEW in ver 3.x) or spatial population expansion
can be estimated
Haplotype frequency estimation
Estimates the frequency of haplotypes present in the
population by maximum likelihood methods.
Gametic phase estimation
(NEW in ver 3.x)
Estimates the most like gametic phase of multi-locus
genotypes using a pseudo-Bayesian approach (ELB algorithm).
Linkage disequilibrium
Test of non-random association of alleles at
different loci.
Hardy-Weinberg equilibrium
Test of non-random association of alleles within
diploid individuals.
Tajima’s neutrality test
Test of the selective neutrality of a random sample
of DNA sequences or RFLP haplotypes under the infinite site model.
Fu's FS
neutrality test
Test of the selective neutrality of a random sample
of DNA sequences or RFLP haplotypes under the infinite site model.
Ewens-Watterson neutrality test
Tests of selective neutrality based on Ewens sampling
theory under the infinite alleles model.
Chakraborty’s amalgamation test
A test of selective neutrality and
population homogeneity. This test can be used when sample heterogeneity is
suspected.
Minimum Spanning Network (MSN)
Computes a Minimum Spanning Tree (MST)
and Network (MSN) among haplotypes. This tree can also be computed for all
the haplotypes found in different populations if activated under the AMOVA
section.
Inter-population methods:
Short description:
Search for shared haplotypes between populations
Comparison of population samples for their haplotypic
content. All the results are then summarized in a table.
AMOVA
Different hierarchical Analyses of Molecular
Variance to evaluate the amount of population genetic structure.
Pairwise genetic distances
FSTbased genetic distances for short divergence time.
Exact test of population differentiation
Test of
non-random distribution of haplotypes into population samples under the
hypothesis of panmixia.
Assignment test of genotypes
Assignment of individual genotypes to particular populations according to
estimated allele frequencies.
Mantel test:
Short description:
Correlations or partial correlations between a set of
2 or 3 matrices
Can be
used to test for the presence of isolation-by-distance
Estimate population specific two-level F-statistics
assuming Hardy-Weinberg equilibrium
Estimate population specific two-level F-statistics
considering inbreeding
Bootstrap across loci to estimate confidence
interval
Phylogenetic analysis
Estimate frequency from DataSet
Estimate distance based Frequency data using 19
different methods
Construct UPGMA tree
Construct NJ tree
Bootstrap across loci to construct multiple trees
for tree consensus
Association study
Allele test
Genotype test
Trend test
Distance test
Exact test
Genotype based F-test
Haplotype trend regression for binary and
quantitative traits
Design
Choose core set of lines by allele number, allelic
diversity, allelic entropy. Selection can be done with simulated annealing,
random search or exhaustive search under general constrains
Choose haplotype tagging markers from haplotype
data
Choose haplotype tagging markers from genotype
data
Choose haplotype tagging markers from trio
data
Tools
Mantel test
Contigency table analysis
SNP identification from sequences
Parse Structure's result
SNP simulation under coalescence model
SNP simulation under coalescence model with
recombination hotspots
아
버지가 아들에게 그리고 그 아들이 자신의 아들에게, 이렇게 아버지에서 아들로 계속 이어지는 것으로는 무엇이 있을까? 아버지를
모른다거나, 입양되지 않은 한, 아들은 아버지로부터 김 씨인지, 이 씨인지, 박 씨인지 등과 같은 성을 물려받는다.
생
물학적으로는 어떨까? 아버지에서 아들에게로만 세대를 거쳐 계속 전달되는 것이 있다. 바로 Y염색체이다. 자식은 각 부모로부터
23개의 염색체를 물려받아 46개의 염색체를 갖는다. 그런데 아버지의 46개 중 아들에게만 넘어가는 것이 Y염색체이다.
Y염색체는 사실상 성보다 더 확실한 부자간의 끈이다. 성은 중간에 바뀌는 일이 생길 수 있으니까 말이다.
Y염색체와 성 간의 공통점
어쨌건 성과 Y염색체는 둘다 아버지에게서 아들에게로만 이어지는 공통점이 있다. 그렇다면 이 점을 이용해 DNA로부터 성을 찾을 수 있지 않을까?
이런 생각이 실제로 가능하다는 소식이 영국의 국영방송 BBC에서 보도되었다. DNA가 이제 친자관계뿐 아니라 성씨까지도 찾아주는 시대가 온 것이다.
패
밀리트리DNA(Family Tree DNA)라는 이름의 유전 검사 회사는 12만5천 명의 남성으로부터 얻은 유전 자료로
Y서치라는 데이터베이스를 구축했다. 이 데이터베이스는 Y염색체에서 특정 성씨가 갖는 유전적인 표지를 무엇인지를 찾아내
만들어졌다.
패밀리트리DNA는 성을 찾고자 하는 고객에게 3가지 DNA 테스트 중 하나를 선택할 수 있도록
했다. 성에 대해 Y염색체에서 12개, 37개 또는 67개의 유전적인 표지를 쓸 것이냐 하는 것이다. 검사에 드는 비용은 각각
149달러, 259달러, 349달러이다.
Y서치를 이용한 유전검사는 지푸라기라도 잡고 싶은 심정의 입양인들에게 도움이 될 수 있다. 부모에 대한 어떤 정보도 갖고 있지 않은 입양인들은 자신의 성이라도 찾기란 매우 어렵기 때문이다.
너무 흔하지도 너무 희귀하지 않은 성이 적합
▲ 남성은 커다란 X염색체 1개와 자그마한 Y염색체 1개를 갖고 있다. 이 Y염색체는 세대를 거쳐 아버지에서 아들에게로만 전달된다.
최근 Y서치를 이용해 자신의 원래 성을 찾고자, 최소 30명의 남성이 이 회사에 등록했다. 실제로 이 데이터베이스를 이용하는 고객은 어린 시절에 입양된 사람들이다.
한 예로, 챈들러 바버라는 37살의 광고카피라이터는 태어났을 때 입양되었다. 그는 Y서치 데이터베이스에서 리치라는 성을 가진 6명과 유전적으로 일치하며, 루에치라는 미국의 성을 가진 한 사람과도 상당히 일치한다는 결과를 받았다.
또
다른 한 예로는, 친부의 성이 페이지라는 사실을 당초부터 알고 있는 48살의 데드워드 세루로의 사례이다. 그는 패밀리트리DNA의
서비스를 통해 자신과 비슷한 성씨 유전자를 가진 22명의 결과를 받았다. 그런데 22명 중 11명의 성이 페이지이었다. Y서치
데이터베이스가 통계적으로 꽤 좋은 결과를 보여준 셈이다.
하지만 Y서치가 모든 성을 잘 찾아줄 수는 없다. 너무 흔하지도 너무 희귀하지도 않은 성이 딱 적당하다고 한다.
따
라서 현재 상황으로 볼때 우리나라처럼 김이박처럼 특정 성씨가 매우 흔한 경우에는 DNA를 이용한 성씨를 찾기란 쉽지 않다.
하지만 김이박에도 여러 파가 나뉘어져 있으니, 어쩌면 미래에는 DNA가 당신은 무슨 성씨에 무슨 파라고 말해줄지도 모른다.
- 잡담 >> 내가 하는 일과 관련된 내용이다보니 재밌다. Y염색체가 아버지를 통해서 남자에게만 유전되고, 성씨도 남자의 것을 물려받으니 가능한 듯 하다. 좋은 아이디어다. 참, 별걸로 다 돈을 벌려고 한다.ㅋㅋ 하지만, 이건 말 그대로 유전적 정보만을 보여주기 때문에, 과거에 조상중에서 다른 집안에서 입양되거나 데려다 키워서 아버지가 다르거나 한 경우에는... 문제가 생길수도 있겠다. 혹은 요즘처럼 어머니의 성씨를 물려받을 수 있는 경우에는... 자신의 원래 아버지쪽이 누구인지 알 수도 있겠지만, 혹은 이런 내용이 고려되지 않게 되면 혼란스러울 수도 있을듯하다. 또, Y염색체를 이용하기 때문에 남자만 가능하다. 이런걸 상업적으로 이용하려면 수많은 DNA데이터베이스를 갖고 분석을 거쳐야 할 것이다. 외국은 범죄자 데이터베이스등에 많은 데이터가 존재하기 때문에 저런 분석이 가능할지 모르지만... 본인 허가없이 DNA 샘플링도 할 수 없고, 간단한 개인정보가 포함된 데이터베이스조차 만드는게 법적으로 금지된 우리나라에서는 아직은 힘든 이야기일 것이다.